This essay is part of The Launch Sequence, a collection of concrete, ambitious ideas to accelerate AI for science and security. These projects need people to build them. Get in touch.
Summary
FDA submissions contain unparalleled detail about the design of clinical trials, manufacturing processes, safety assessments, and reviewer correspondence. Yet these documents submitted to the FDA are not publicly available. This gives a structural advantage to large pharmaceutical incumbents who have amassed regulatory filing archives over small biotechs. As private regulatory archives and institutional know-how are costly to replicate, new innovators are effectively boxed out. Meanwhile, AI stands ready to revolutionize drug development by auto-drafting submissions, predicting approval outcomes, and optimizing trial protocols — but only if it can be trained on unredacted, high-quality datasets.
This proposal outlines a bold, legally grounded mechanism to democratize FDA filings: leveraging existing US bankruptcy law to create an open-source library of orphaned FDA submissions (specifically, Investigational New Drug applications, New Drug Applications, and Biologics License Applications) and anonymized clinical trial results from failed drug sponsors. Freed from obscurity, these documents will power AI-driven regulatory intelligence tools that dramatically lower compliance costs, accelerate approval timelines, and level the playing field for small biotech firms, academic teams, and nonprofits — ultimately delivering novel therapies to patients faster and more affordably.
Motivation
The US can’t afford to lose the biotech race
In the global biotech race, the United States is losing ground fast, and the consequences could be profound. A Time magazine headline from May 2025 captures what many industry experts have been warning for years: “The US can’t afford to lose the biotech race with China.” This is no longer a hypothetical concern. Data is backing this fear up: US early-stage funding is deteriorating: dropping from $2.6 billion in Q1 to just $900 million in Q2 2025 — the lowest level in five quarters.
Meanwhile, China designated biotechnology a strategic industry in 2011 and streamlined its drug development regulations in 2015. It has since committed tens of billions of dollars to R&D, overtaking the United States in the number of active clinical trials, and is becoming a dominant source of globally licensed drug candidates. A key structural advantage for Chinese startups is a more flexible regulatory system and faster path to clinical rollout. Chinese start-ups are reportedly entering clinical trials within 18 months of founding, compared to several years for their US counterparts.
Innovation is increasingly driven by start-ups — but regulation favors large incumbents
To remain competitive, US biotech startups need faster, more efficient ways to engage with the regulatory system. Multiple independent analyses show that in the past decades there has been a shift in the biotech innovation landscape, with small and mid-sized biopharma now leading the way in discovering and advancing innovative new drugs, as opposed to large biopharmaceutical companies.
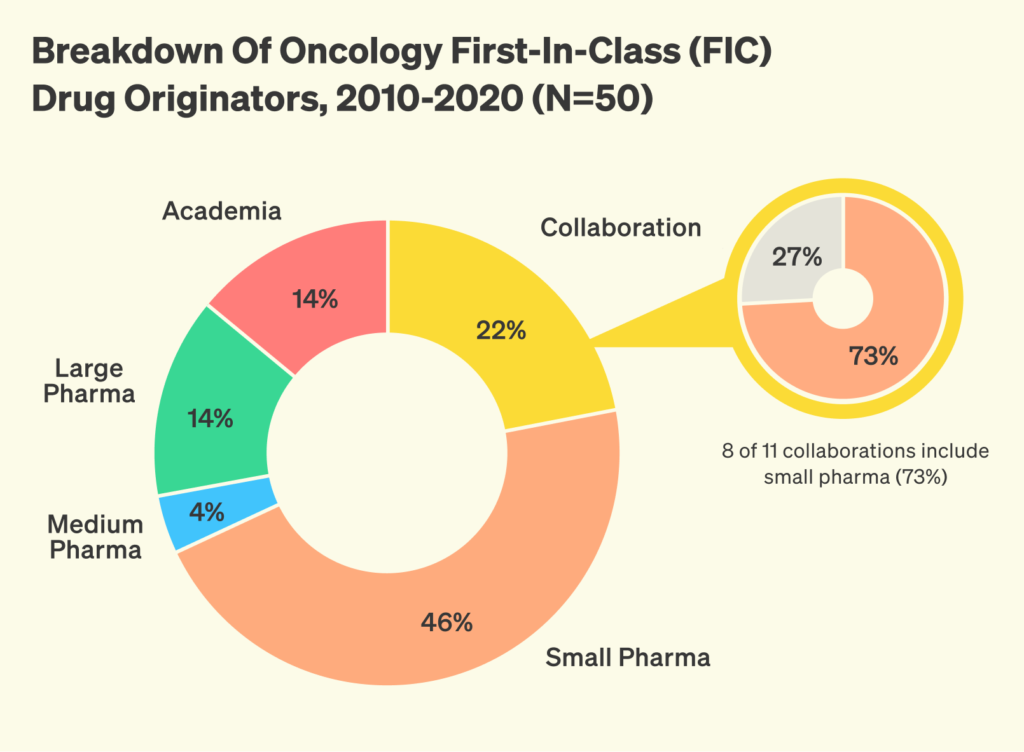
But these smaller players face significant regulatory barriers that larger incumbents are better equipped to navigate. Small teams lack the internal regulatory teams, prior FDA correspondence, and historical filing libraries that help incumbents move quickly. As a result, they often rely on expensive consultants, or trial and error. In informal surveys with biotech startup founders, many report difficulty interpreting precedent, a desire for greater access to historical filings, and inconsistent guidance from outside advisors.
Brian Finrow, CEO of start-up LumenBio, explains how access to prior Investigational New Drug (IND) filings would derisk the development of new technologies: “Our company is developing a new class of biopharmaceuticals that could offer major advantages over traditional GMP manufacturing. However, the FDA expects regulatory submissions to follow conventional formats — but formal guidance offers little clarity for such novel technologies. While consultants provide some guidance, they often lack deep familiarity with our specific technology, creating a Catch-22. Even limited open access to prior IND filings would significantly accelerate development and reduce risk for innovators working on first-in-class therapeutics.”
These informal surveys are confirmed by empirical results from the economics of innovation. When it comes to the biopharmaceutical industry, regulatory complexity favors large incumbents. A landmark analysis of 766 new molecular entities submitted to the FDA between 1979 and 2000 found that between 30–55% of the advantage enjoyed by large firms in approval timelines can be attributed to familiarity with the regulatory processes alone. A recent analysis of FDA medical device deregulation between 1980 and 2015 found that reclassifying certain devices from Class II to the lower scrutiny Class I led to both higher quality and quantity of innovation, without compromising safety. Notably, smaller firms saw the greatest benefit, with new firm entry increasing by 200%. The author attributes this to reduced approval delays and a flatter regulatory learning curve.
The FDA sits on a treasure trove of data
An overlooked opportunity to address this challenge lies in combining AI with expanded access to historical FDA regulatory filings, which are currently largely unavailable, to create powerful tools that could act as “regulatory co-pilots.”
Submissions to the FDA for drug approval are known as Common Technical Documents (CTDs), following an internationally standardized format. In their mature form, a New Drug Application (NDA) for small molecules or a Biologics License Application (BLA) for biologics, they cover every aspect of a new drug’s development, including administrative data and labeling, detailed manufacturing methods, animal study data, clinical trial results, and communications with the FDA. CTD dossiers often span 10–20,000 pages; collectively, they form one of the most exhaustive repositories of real-world scientific practice and regulatory negotiation ever assembled, and thus a rich resource for AI to be trained on. However, this information is very hard to access. Bound by statutory law,1 the FDA treats most of it as confidential, especially anything seen as a trade secret or business-sensitive. In practice, this results in heavily redacted FOIA releases that lack the technical detail that small sponsors would require.
Many start-up founders have said it would be particularly valuable to access the Chemistry, Manufacturing and Controls (CMC) part of CTD documents. Collectively, CMC data describe how a drug product is made, tested, and maintained to ensure its quality, safety, and consistency. CMC alone represents 13–17% of total R&D expenditures, yet demands specialized process chemistry and analytical know-how that small biotechs often lack, often leading to the need to hire expensive consultants. This is also the part that is currently hardest to obtain under the FOIA exemption, as it is most likely to contain information that can be classified as a trade secret.
AI could unlock the value — if we unlock the data
The importance of democratized access to past filings has sharply increased with the rise of AI. These tools have the power to dramatically streamline the regulatory process. This has been recognized by the FDA itself, which has decided to incorporate AI into its decision-making process with the goal of accelerating review timelines and standardizing decisions. Academic papers confirm the feasibility of such an approach and suggest it could save billions in regulatory costs. But so far, these efforts are limited to internal use; sponsors do not have access to similar tools to improve their own submissions. Meanwhile, large pharmaceutical companies are also building internal AI systems to help navigate the regulatory process, making use of their large repositories of CTDs and clinical trial results. As a result, AI’s promise to make the regulatory processes more efficient may widen the gap between large and small biotech firms. Without broader access to the underlying data needed to train effective AI tools, smaller companies risk being left behind in an increasingly data-driven regulatory environment.
A specific use case for AI-driven natural language processing is interpreting complex sections of FDA filings and converting them into standardized templates. Such templates could accelerate the drafting of new submissions and ensure that critical regulatory precedents are accurately reflected in every document. For example, the large pharmaceutical company Novo Nordisk used its own internal database of clinical study reports to train a purpose-built machine-learning model for automating the drafting of these documents. Digitalization Strategy Lead Waheed Jowiya declares that: “We’ve reduced the time taken to create Clinical Study Reports from 12 weeks to 10 minutes, with higher quality outputs and a fraction of the team. In terms of value, each day sooner a medicine gets to market can add around $15 million in revenue to the company.”2
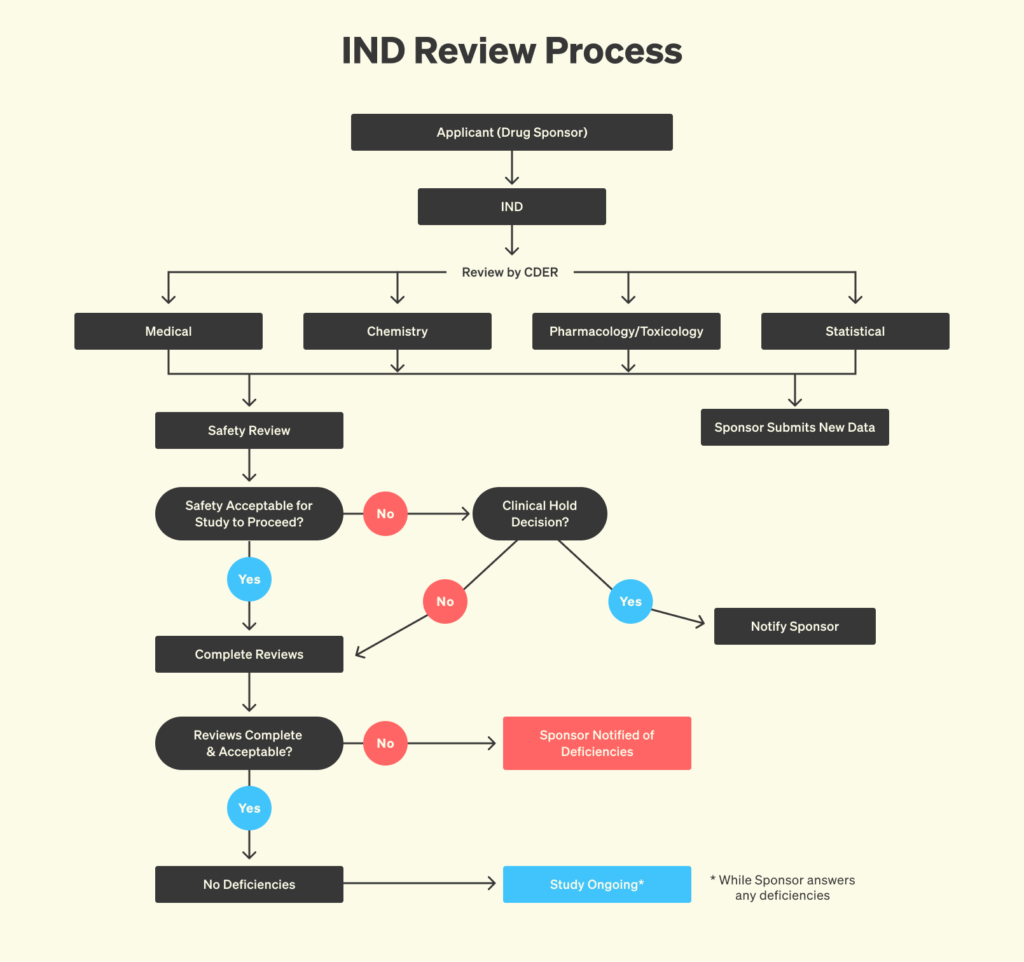
Beyond regulatory document automation, AI could offer powerful predictive insights into regulatory outcomes. By analyzing historical data on review cycles, amendment requests, and final decisions, AI models can identify patterns that correlate with successful approvals or common reasons for rejection. This would allow sponsors to forecast the likelihood of clearance at each stage of the review process, prioritize resources on the most promising development pathways, and proactively address potential concerns. Early detection of red flags, such as insufficient toxicology data or inconsistent batch validation protocols, would enable project teams to adjust study designs or manufacturing plans before they become obstacles, ultimately minimizing costly delays and rework.
This is an opportunity for policy and philanthropy
The market won’t fix this on its own. Most CTDs, even from failed drugs, remain locked behind FOIA exemptions indefinitely. This is a classic coordination problem: no individual company has an incentive to release data, but the collective value of shared access could be transformative.
A thoughtful balance must be struck. Despite the clear benefits outlined above, indiscriminate disclosure of complete CTDs, especially of actively developed assets, would risk undermining the very incentives that drive pharmaceutical innovation, which is already a high-risk endeavour. However, according to a comprehensive 2020 study, almost half of US biotech ventures fail within five years and a quarter become “walking zombies” with no meaningful activity. This means that we are sitting on a vast archive of sunk-cost scientific knowledge that could be repurposed. Making use of CTDs from these failed companies could enable an AI-driven regulatory renaissance without compromising the commercial potential of any given enterprise.
Solution
Currently, the FDA operates under FOIA Exemption 4 and is exposed to litigation risk from drug sponsors if it discloses information deemed confidential. Under current law, the FDA must notify companies prior to any potential release and honor their objections, unless it can prove that the disclosure would not cause substantial competitive harm. As a result, the agency tends to interpret “trade secrets” broadly and defaults to withholding information. Currently, even CTDs for assets that are 20 years old and have been discontinued remain inaccessible in full, due to overbroad confidentiality protections. One potential solution is to amend the legal definition of a “trade secret” and allow for greater public access to regulatory filings, such as permitting the release of historical CTD dossiers for drugs that have been developed more than a number of decades ago and whose patents have expired.
However, such reform is procedurally complex and politically uncertain. Furthermore, it may not go far enough to address the need for transparency in newer scientific modalities, such as cell therapies, gene editing, or mRNA platforms. A more immediate and pragmatic alternative that bypasses the burdens of legal change and offers access to CTDs covering newer technologies is to establish an AI Regulatory Fund dedicated to acquiring the regulatory dossiers of the biotech companies that have entered bankruptcy. Capitalized through federal funding and/or philanthropic support, the Fund will assemble a small team of legal experts and data engineers to monitor bankruptcy cases involving biotechnology sponsors. When a company’s IND, NDA, or BLA assets risk falling into obscurity, the Fund will submit calibrated offers to secure non‐exclusive rights to this information.
This approach has been used successfully in the past at a smaller scale. In one recent biotech bankruptcy case, two entire CTD files were transferred for $25,000 apiece. Extrapolating from that precedent, we estimate that assembling a foundational library of 20 CTDs across each of five major drug modalities (peptide, antibody, small molecule, gene therapy, and cell therapy) would require 100 dossiers in total, at $25,000 each, for a program cost of approximately $2.5 million. This is a very low cost compared to the cost of generating such data, which can range from a few million for an IND to hundreds of millions for an NDA/BLA. Given that individual CTDs often span thousands of pages of detailed chemistry, manufacturing, controls, preclinical, and clinical data, even this relatively modest collection would offer valuable insights. However, scaling to several hundred or more submissions would unlock exponentially greater returns in predictive power and regulatory pattern discovery.
Upon acquisition, each dossier will be digitized in its entirety, with personal identifiers removed in accordance with privacy standards, ensuring that this process preserves scientific reasoning and regulatory correspondence. The files will then be ingested into a secure, cloud‐based repository, complete with full‐text search and chronological indexing. This can be modelled on the EDGAR system (Electronic Data Gathering, Analysis, and Retrieval), a public platform that collects and publishes filings from companies required to report to the US Securities and Exchange Commission. It enables investors, analysts, and regulators to access corporate disclosures such as financial statements, risk factors, and executive compensation in a searchable, structured format. EDGAR promotes transparency, accountability, and market efficiency by making regulatory information freely and systematically available.
With this corpus in place, developers can train a specialized language model that understands the FDA’s implicit decision criteria. Researchers drafting a new IND could query the model in natural language and receive precise, context‐sensitive guidance on assay validation strategies or study‐design considerations. Project teams would be able to forecast likely regulatory concerns, draft first‐pass submission documents that align with reviewer expectations, and reduce the number of costly amendment cycles.
This initiative operates entirely within existing legal frameworks. Under federal law, bankruptcy court judges have broad powers to transfer title to any asset owned by the failed company, including rights to data and information included in FDA filings. This authority is granted under 11 U.S.C. § 363, which allows courts to approve the sale of both tangible and intangible assets, including intellectual property and regulatory submissions. Importantly, no involvement of the FDA as a disclosing party is required, avoiding conflict with FOIA Exemption 4 and trade secret protections. By acting as a benign bidder, the Regulatory Transparency Fund ensures that any entity with a credible plan to revive a failed drug program can outbid it, preserving incentives for the continued development of any particular drug candidate. Meanwhile, small firms, academic researchers, and non-profits would gain a lasting infrastructure for regulatory know‐how.
The AI Regulatory Transparency Fund thus represents an ambitious but practical undertaking: it requires modest resources compared to its impact, and bypasses the complexity and lengthy timelines of changing laws, while holding the potential to catalyze an AI‐driven renaissance in regulatory science and level the competitive playing field for the small innovators who will bring tomorrow’s therapies to patients.
Further resources
- Alex Telford, Will all of our drugs come from China?, 2024.
- The Economist, It’s not just AI. China’s medicines are surprising the world, too. February 2025.
- Parker Rogers, Regulating the Innovators, November 2023.
- Enlli Lewis, Not all clinical trial data in the US are fragmented, June 2025
- Joshua M. Sharfstein, Michael Stebbins, Enhancing Transparency at the US Food and Drug Administration, April 2017.
- Stuart Buck, Improving FDA transparency for public health, November 2024.
- Eric Budish, Benjamin Roin, Heidi Williams, Do Firms Underinvest in Long-Term Research? Evidence from Cancer Clinical Trials, 2015.
- Joshua Jackson, Georgina Jones Suzuki, Zoe Dettelbach, David M. McIntosh, Protecting trade secrets from FOIA disclosure in the Wake of FDA lay-offs, May 2025.
Appendix
Additional context on CTDs
Submissions to the FDA follow the Common Technical Document (CTD) framework, an internationally standardized filing system that covers every aspect of a new drug’s development, including administrative data and labeling, detailed manufacturing methods, animal study data, and clinical trial results. The document starts life as an Investigational New Drug (IND) application, which focuses especially on manufacturing plans, animal toxicology findings, and protocols for initial human trials. As the drug candidate progresses through clinical trials, more and more information is added to the CTD dossier. The CTD also includes a written record of most interactions between the FDA and the drug sponsor throughout development. When the drug sponsor believes that the data support approval, that same CTD dossier evolves into a New Drug Application (NDA) for small-molecule drugs or Biologics License Application (BLA) for vaccines, cell therapies, and other biologic drugs, enriched with comprehensive clinical data, final validations, and proposed labeling.
The current state of obtaining information under FOIA and the legal landscape
Accessing abandoned Investigational New Drug (IND) applications through the Freedom of Information Act (FOIA) is rare and challenging — especially when it comes to Chemistry, Manufacturing, and Controls (CMC) data. In a 2019 article, researchers describe the process through which they sought clinical safety data on Ro 24-7429, an old HIV drug from Hoffmann-La Roche, for repurposing in leukemia. Despite over 20 years having passed since the IND was active and the sponsor having publicly announced the discontinuation of the program, the FDA still redacted all CMC-related content. FDA staff made clear that the barrier for releasing CMC data is significantly higher than for safety or efficacy data. Ultimately, the released 464 pages included only non-CMC materials — pharmacology reviews, clinical protocols, and adverse event data — underscoring that even for long-abandoned programs, CMC information remains nearly inaccessible under FOIA.
The legal landscape of what constitutes a trade secret is complex. Historically, courts applied a standard from the 1974 National Parks decision, which requires agencies to demonstrate that disclosing confidential information would cause substantial competitive harm to the company that submitted it. This placed a real burden on the agency and created a check on over-withholding. But in 2019, the Supreme Court significantly changed the legal landscape with its decision in Food Marketing Institute v. Argus Leader Media. The Court rejected the competitive harm test and redefined “confidential” to simply mean information that is customarily kept private and is shared with the expectation it will remain secret. This decision made it much easier for the FDA to justify withholding information under Exemption 4, even when the competitive consequences of disclosure are unclear or minimal. The 2016 FOIA Improvement Act complicates the picture by requiring agencies to show a “foreseeable harm” before withholding information under any FOIA exemption. In Seife vs FDA 2022, the Second Circuit addressed this tension directly. The court upheld the FDA’s decision to withhold confidential information related to a drug approval and interpreted the “foreseeable harm” requirement to mean harm to the commercial or financial interests of the information’s submitter, or harm to the government’s interest in maintaining confidentiality and continued voluntary cooperation from private entities.
How bankruptcy law enables the transfer of intellectual assets
Under existing U.S. federal law, bankruptcy courts have broad authority to transfer ownership of a failed company’s assets, including rights to data and regulatory filings such as FDA INDs, NDAs, and BLAs. This authority is granted under 11 U.S.C. § 363, which allows courts to approve the sale of both tangible and intangible assets, including intellectual property and regulatory submissions. Bankruptcy courts have repeatedly approved such transfers in practice. These transactions are often handled through court-approved Asset Purchase Agreements (APAs), which can be very simple. While the FDA itself does not transfer ownership, it recognizes the new owner once proper documentation (e.g., transfer letters) is submitted, as outlined in 21 CFR § 314.72 (for NDAs) and 21 CFR § 601.72 (for BLAs).
-
Freedom of Information Act’s (FOIA) Exemption 4 (5 U.S.C. § 552(b)(4)), codified at 21 CFR §20.6.
-
These high numbers can be explained by the fact that patent exclusivity is granted from the date of filing of the patent, whereas commercialization starts from the time the drug is approved, so any delay in the regulatory process cuts from commercialization time. While most drugs fail, those that do make it through can create a lot of revenue: for example, Humira generated $21.2 billion in revenue for Abbvie in 2022 alone. The incentives created by this way of granting patent exclusivity are explained in a 2015 paper from economist Heidi Williams.
